Random sampling from genomic sequences
random_select_multi.py - Sample random sequences from a genome
random_select_multi.py code and install instructions can be found on the GitHub page: https://github.com/Daniel-Ze/python_scripts/tree/main/random_select_multi
Why you would sample random sequences from a genome
I wrote this script for the analysis of random sequence capture events of RC/helitrons in genomes. RC/helitrons are class II transposable elements and therefore belong to the DNA transposons. The special thing about these retroelements is their rolling-circle replication mechanism and the associated possibility of gene capture. It is known for other transposable elements to be able to capture genes causing gene duplications or knockouts by insertion in the coding sequence influencing morphological traits. One good example would be the LTR retrotransposon Rider found in tomato. This LTR retroelement family was shown to have expanded exclusively in the genome of tomato. Phenotypes influenced by Rider are for example fruit shape and jointlessness. These are caused by either retro-duplication of the SUN gene or the knock-out of a SEPALATA MADS-box gene by retrotransposon-insertion into the coding sequnce [1 ,2].
RC/helitrons were first discribed in the genome of bats. This type of transposon was shown to have heavyly spread throughout these genomes making up around 6% of the Little Brown Bat (Myotis lucifugus) genome [3]. Their replication mechanism was shown to be able to capture partial or even complete genes allowing them to transpose to novel positions in the genome. The same was shown to happen also in the genome of maize where autonomous RC/helitrons were identified. They have the ability to "paste" themselves together with captured gene fragments into novel positions [4].
Assuming now that these retroelements contribute to the diversification of a certain gene family we would expect an enrichment of the gene family of interest among the retroelement sequences. We can extract the retroelement-associated sequences and the encapsulated genomic sequences and check for blast similarities to our gene family of interest or serch for conserved domains via hidden markov models (HMMs). To confirm an enrichment we can now draw random samples from the genome. Running the same analyses as for the retroelement associated sequences with these randomly sampled sequences, you should get some insight into possible enrichment or not.
How to sample random sequences from a genome with random_select_multi.py
Before we start randomly drawing samples from a genome we need to go through a checklist first. There are a few things we might want to consider:
- How many random sequences do we want to draw
- What size range should the random sequences represent
- How often should the random sequence sampling be repeated
Obtaining paramters and running the script
For the first two points we can orient ourselves on the extracted transposon sequences. How many are there and how is the size distribution of the sequences?
# 1. Count the sequences:
(base) server user ~ $ grep -E '^>' transposon.fasta | wc -l
# 2. Get min and max for sequence length range:
# - first count the bp of each sequence:
(base) server user ~ $ awk '/^>/ {if (seqlen){print seqlen}; print ;seqlen=0;next; } { seqlen += length($0)} END{print seqlen}' transposon.fasta > transposon_seqlen.txt
# - print the first and last line of list:
(base) server user ~ $ grep -v '^>' transcripts_seqlen.txt | sort -n | awk 'NR == 1 { print }END{ print }'
For point number 3 we can chose for example 100 repetitions. This will give us enough variation to determine if there is significant enrichment in our transposon sequences.
# 1. Count the sequences:
(base) server user ~ $ grep -E '^>' transposon.fasta | wc -l
# 2. Get min and max for sequence length range:
# - first count the bp of each sequence:
(base) server user ~ $ awk '/^>/ {if (seqlen){print seqlen}; print ;seqlen=0;next; } { seqlen += length($0)} END{print seqlen}' transposon.fasta > transposon_seqlen.txt
# - print the first and last line of list:
(base) server user ~ $ grep -v '^>' transcripts_seqlen.txt | sort -n | awk 'NR == 1 { print }END{ print }'As we have determined all the important facts we can head over to random_select_multi.py and have a look at the required input options:
# Dry run of random_select_multi.py:
(base) server user ~ $ python random_select_multi.py
Usage: python random_select_multi.py -i fasta.fai -n 100 -s 300 -r 200:20000 -c 10
-i / --input= Index of fasta file e.g. Seq1 199192370
-n / --nsample= Number of times to draw random samples (default 100)
-s / --nseqs= Number of random sequences to draw each time
-r / --range= Size range of random sampled sequences
-c / --cpus= Number CPUs to use- -i / --input= We need to give the script a index file with the start and stop coordindate of each chromosome / contig / scaffold etc.
- -n / --nsample= Here we supply the 100 repetitions that we decided on earlier.
- -s / --nseqs= Here we supply the identified number from point 1 earlier.
- -r / --range= Here we supply the length range of the sequences from point 2.
- -c / --cpus= Here you can supply the number of CPUs that you can use for the analysis.
awk '/>/{if (l!="") print l; print; l=0; next} {l+=length($0)} END {print l}' genome.fa | paste - - | cut -d ">" -f2 > genome.indexWhat is the script doing in detail
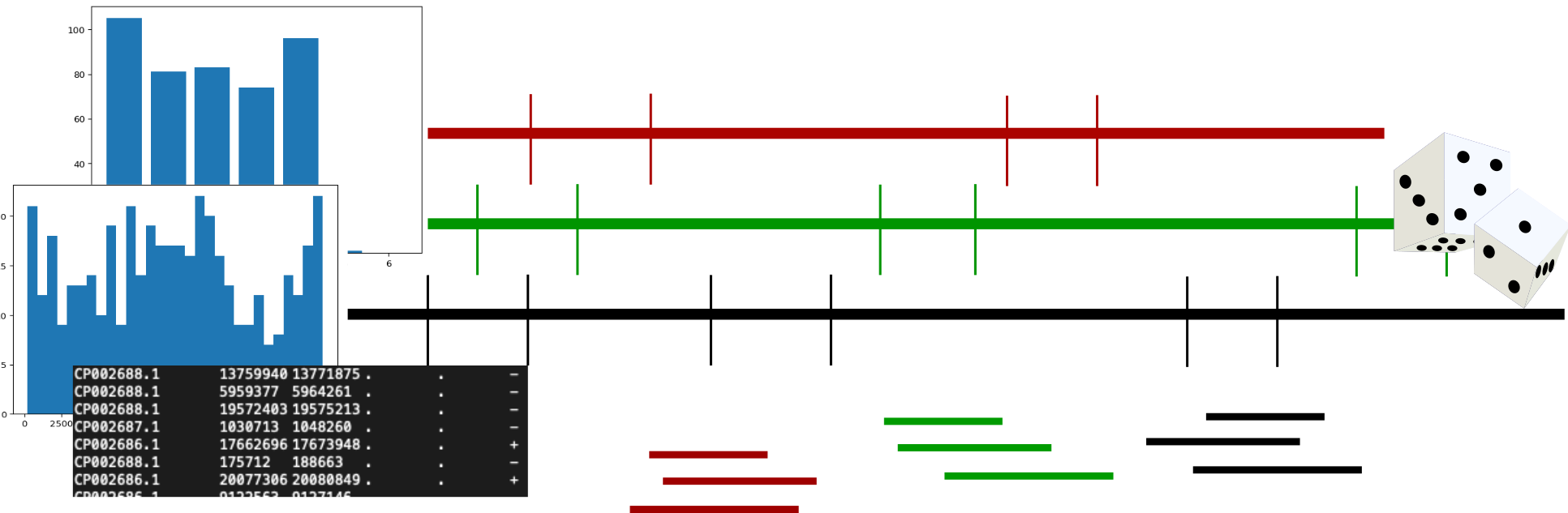
The detailed steps of the script are described in the following flow-chart:
 As you will be dealing with genomes of variable quality, in terms of completeness and continuity, the first step of the script is to calculate the relative size of each contig / scaffold / chromosome. This will be used as sampling probability: larger sequence chunk = more likely to randomly sample from it. After this, the script generates a list of sequence lengths in the supplied length range. It is using a equal distribution for each size range. After this the actual random sampling starts. First a random sequence from the list of sequences is chosen. Next a random start cordinate is chosen for the sequence length in the list. Next the stop cordinate is calculated according to the sequence length plus start coordinate. If this is out of bounds of the chosen sequence the script tries three times to sample a new start coordiante before it samples a new sequence and restarts the whole process. If the script finds a fitting start and stop coordinate it continues with the next random sequence length until all sequences are sampled. This process is then repeated n times for the repetitions. These random samplings are executed in parallel as long as the user has supplied a number greater than 1 for the argument -c / --cpus=.
As you will be dealing with genomes of variable quality, in terms of completeness and continuity, the first step of the script is to calculate the relative size of each contig / scaffold / chromosome. This will be used as sampling probability: larger sequence chunk = more likely to randomly sample from it. After this, the script generates a list of sequence lengths in the supplied length range. It is using a equal distribution for each size range. After this the actual random sampling starts. First a random sequence from the list of sequences is chosen. Next a random start cordinate is chosen for the sequence length in the list. Next the stop cordinate is calculated according to the sequence length plus start coordinate. If this is out of bounds of the chosen sequence the script tries three times to sample a new start coordiante before it samples a new sequence and restarts the whole process. If the script finds a fitting start and stop coordinate it continues with the next random sequence length until all sequences are sampled. This process is then repeated n times for the repetitions. These random samplings are executed in parallel as long as the user has supplied a number greater than 1 for the argument -c / --cpus=.
Having a look at the results
The script produces several outputs:
- Barplot of number of sequences sampled from different chromosomes / contigs / scaffolds
- Histogram of sequence length distribution of random sequneces
- Histogram of randomly chosen start positions
- .bed files with coordinates of the randomly sampled sequences
- .fasta files with the actual sequences of the random coordinates
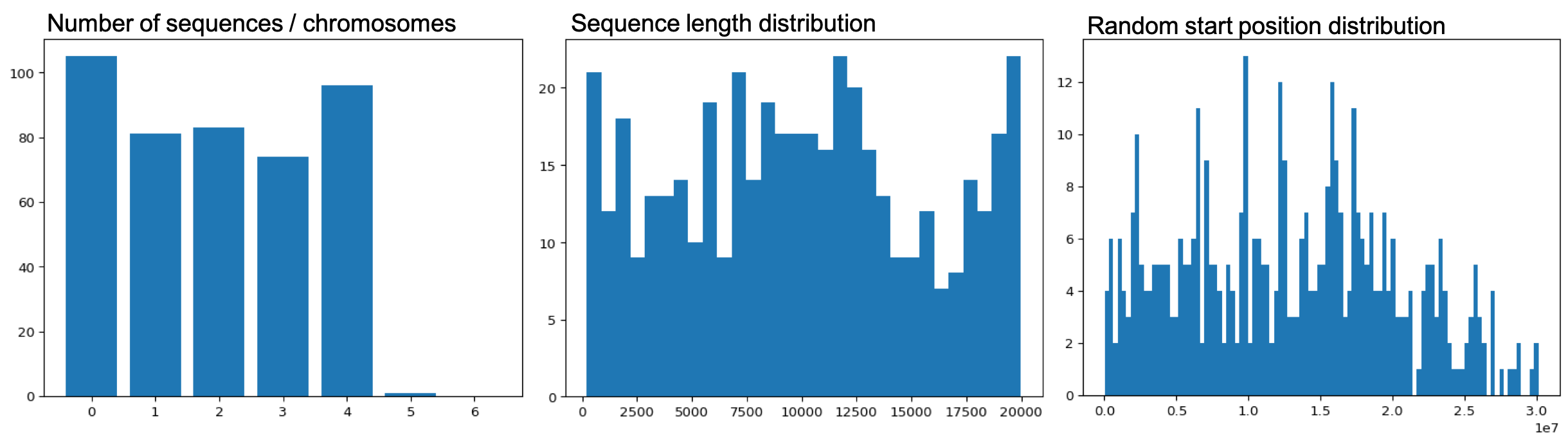 These plots plus .bed and .fa files will be created in a folder called random_sampling within the folder with the genome file. The .fa files can then be used for further analysis.
These plots plus .bed and .fa files will be created in a folder called random_sampling within the folder with the genome file. The .fa files can then be used for further analysis.
References
1. Xiao, H.; Jiang, N.; Schaffner, E.; Stockinger, E.J.; Van Der Knaap, E. A retrotransposon-mediated gene duplication underlies morphological variation of tomato fruit. Science (80-. ). 2008, 319, 1527–1530, doi:10.1126/science.1153040.
2. Soyk, S.; Lemmon, Z.H.; Oved, M.; Fisher, J.; Liberatore, K.L.; Park, S.J.; Goren, A.; Jiang, K.; Ramos, A.; van der Knaap, E.; et al. Bypassing Negative Epistasis on Yield in Tomato Imposed by a Domestication Gene. Cell 2017, 169, 1142-1155.e12, doi:10.1016/j.cell.2017.04.032.
3. Thomas, J.; Phillips, C.D.; Baker, R.J.; Pritham, E.J. Rolling-circle transposons catalyze genomic innovation in a mammalian lineage. Genome Biol. Evol. 2014, 6, 2595–2610, doi:10.1093/gbe/evu204.
4. Morgante, M.; Brunner, S.; Pea, G.; Fengler, K.; Zuccolo, A.; Rafalski, A. Gene duplication and exon shuffling by helitron-like transposons generate intraspecies diversity in maize. Nat. Genet. 2005, 37, 997–1002, doi:10.1038/ng1615.